Paroxysmal neurological disorders include a broad spectrum of clinical entities such as migraine, episodic ataxia or myotonia. The research group is focussed on the clinical genetics of epilepsies and paroxysmal dyskinesias, paroxysmal neurological disorders with overlapping clinical and pathophysiological features.
Epilepsy is a very common neurological disease with a life time incidence of up to 3% in the general population. Epilepsies are divided in focal and generalized forms as well as in lesional (induced by e.g. scars, dysplasias or strokes) and genetic (idiopathic) forms looking from a pathophysiological point of view. Up to 30% of epilepsies are genetically determined but only 1-2% are monogenic with a single mutation leading to the clinical syndrome. Most of the genetic epilepsies follow a complex-genetic trait in which only a combination of genetic changes lead to the phenotype.
The genetic epilepsies can be divided in the
(i) idiopathic generalized epilepsies (IGE) like the absence epilepsies with childhood (CAE) and juvenile onset (JAE)
(ii) the idiopathic focal epilepsies such as the nocturnal frontal lobe epilepsy (ADNFLE) or the familial temporal lobe epilepsies (TLE)
(iii) the benign syndromes of early childhood such as the benign familial neonatal (BFNS), infantile (BFIS) and the infantile-neonatal (BFINS) forms and
(iv) the epileptic encephalopathies including the fever-associated syndromes like the generalized epilepsy with febrile seizures plus (GEFS+), the severe form of GEFS+, the Dravet Syndrome (SMEI, severe myoclonic epilepsy of infancy) or the Othahara syndrome.
Epilepsies show a close overlap to the paroxysmal dyskinesias (PD) since the diseases can be found in the same cohort and can be based on the same genetic defect. Paroxysmal dyskinesias can be of lesional origin (e.g. multiple sclerosis lesions found in the basal ganglia) but most of the described cases are of idiopathic/genetic origin. The genetic forms are divided in the following subtypes:
(i) the non-kinesigenic dyskinesia (PNKD)
(ii) the kinesigenic dysinesia (PKD)
(iii) the exertion-induced dyskinesia (PED)
The observed episodic dyskinesias include choreatic, ballistic, athetotic and dystonic features. They can be induced by different stimuli: In PNKD by e.g. stress or alcohol but are not induced by movements; in PKD, the dyskinesias are induced by sudden voluntary movements and last for seconds, and in PED, the dyskinesias appear after a voluntary movement in the exerted muscle group for 30-60 minutes for example in the arms after a heavy arm work.
All PD can be associated to epilepsy in the same patients. PNKD show an overlap to the idiopathic generalized epilepsies with mutations found in the potassium channel gene KCNMA1, PKD with the syndrome of benign familial infantile seizures (BFIS) with mutations found in PRRT2 (proline rich transmembrane protein) and PED to different forms of absence epilepsies with mutations found in SLC2A1 coding for the glucose transporter type 1 (Glut1).
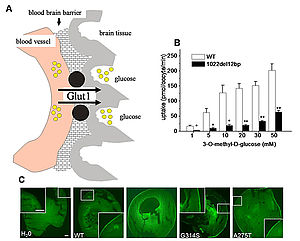
Figure 1: The glucose transporter typ 1 (Glut1)
Glut1 is the most important glucose transporter of the brain since it delivers the glucose over the blood brain barrier (A). Glucose uptake measurements in Xenospus laevis oocytes show a reduced maximum velocity in mutatants compared to wildtype (B). Immcytological studies demonstrate an unchanged surface expression of the transporter in the oocytes (C) (Weber et al. 2008).
Glut1 is the most relevant glucose transporter of the brain since it delivers glucose over the blood brain barrier (figure 1). The Glut1 syndromes include a variety of clinical features with a huge variability in phenotypes. The first mutations in PED were described in 2008. The mutations lead to a functionally relevant reduction in the glucose uptake which can well explain the episodic character of the symptoms under exertion (Weber et al. 2008). Mutations in SLC2A1 were also found in early-onset absence epilepsy (EOAE, Suls et al. 2009) and childhood absence epilepsy (CAE, Striano et al. 2012). All Glut1 syndromes respond well to a ketogenic diet which bypasses the defective glucose metabolism by providing ketones.
The syndrome of benign familial infantile seizures (BFIS) is characterized by a cluster of seizures with an onset between 3 and 12 months of age. The seizures can be treated well by common antiepileptic medications or disappear spontaneously. Most of the patients never develop seizures in adult ages and have a normal psychomotor development. Up to 40% of patients also suffer from PKD which can be also treated by common antiepileptic drugs. Since many years, it was clear that both syndromes are linked to a huge region on chromosome 16 (Weber et al. 2006, Weber et al. 2008) but an underlying gene could not be detected for a long time. Recently, mutations in the gene PRRT2 were described (Schubert et al. 2012, Becker et al. 2013). The resulting protein might be functionally relevant in the vesicle synaptic metabolism of neurons (figure 2).
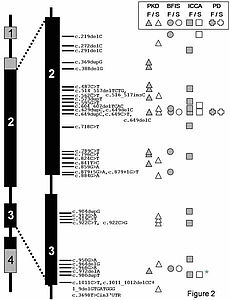
Figure 2: Overview of the mutations found in PRRT2 (Becker et al. 2013)
PRRT2 consists of 4 exons but only the black areas are coding regions. Mutations were found in paroxysmal kinesigenic dyskinesia (PKD), benign familial infantile seizures (BFIS), paroxysmal kinesigenic dyskinesia with infantile convulsions (ICCA) as well as other phenotypes such as paroxysmal non-kineisgenic dyskinesia (PNKD) and paroxysmal exercised induced Dyskinesia (PED) which are summarized as PD.
Recently, a novel gene could be described for BFIS and PKD, SCN8A a sodium channel subunit gene (Gardella et al. 2016).
An additional focus of the working group is the genetic and clinical analysis of epileptic encephalopathies (EE). EEs are early onset, pharmaco-resistant epilepsies which are associated to a psychomotor disturbance. The research group was involved in the description of novel genes for EE such as STX1B, CHD2, GRIN2A, DNM1 and KCNA2 (Suls et al. 2013, Lemke et al. 2013, Schubert et al. 2014, Syrbe et al. 2015, EuroEpinomics-RES consortium et al. 2017).
Additionally, Prof. Weber founded together with Dr. von Spiczak (University of Kiel) the German Commission for Genetics and Epilepsy of the German Society for Epileptology (DGfE) and published guideline for pathophysiology-triggered therapy in epilepsy (see homepage DGfE).

Center of Neurology
Hertie Institute for Clinical Brain Research
Department Neurology and Epileptology
Hoppe-Seyler-Straße 3
72072 Tübingen
Phone: +49 (0)7071 29-80443 or
Phone: +49 (0)7071 29-82048
Fax: +49 (0)7071 29-4488


